RNA-Seq
Overview
RNA-Seq is a next-generation sequencing (NGS) application that analyzes the transcriptome, or every transcript being expressed at a given point in time. Gene expression analysis allow the comparison between two conditions, such as healthy and diseased states. RNA-Seq can also be used quantitatively to determine absolute quantity of each transcript.
General Workflow
A typical RNA-Seq experimental workflow involves the isolation of RNA from samples of interest, generation of sequencing libraries, and use of a high-throughput sequencer to produce hundreds of millions of short single or paired-end reads. There are several variations of the experimental protocol:
mRNA-Seq vs. Whole Transcriptome
Researchers are usually interested in the expression of mRNA, but up to 95% of cellular RNA is rRNA. To avoid wasting reads on rRNA, you can use a selection or a depletion approach.
Selection
For mRNA-Seq, polyA+ RNAs are selected using oligo-dT beads. This excludes rRNAs, most smRNAs, some mRNAs that are not polyadenylated (e.g. histone mRNAs), and nascent mRNAs that have not been fully processed and polyadenylated.
Depletion
Alternatively, for whole transcriptome sequencing, rRNAs can be removed by hybridization to rRNA-specific LNA probes with kits like Ribo-minus/Ribo-zero. This is less efficient, but keeps other RNAs, including unprocessed RNAs in the mixture. some non-mRNA and mRNAs that have not been fully processed (e.g., with introns) appear in the results.
Strand-Specific (aka dUTP) vs. Non-Strand-Specific
To distinguish sense and antisense expression, the dUTP method is used to determine the strand from which the signal comes in RNA-Seq. With this method, the reads will map to the strand opposite to the RNA. Another strand-specific method is based on Smarter approach from Takara. In this case, reads are mapped to the same strand as the RNA.
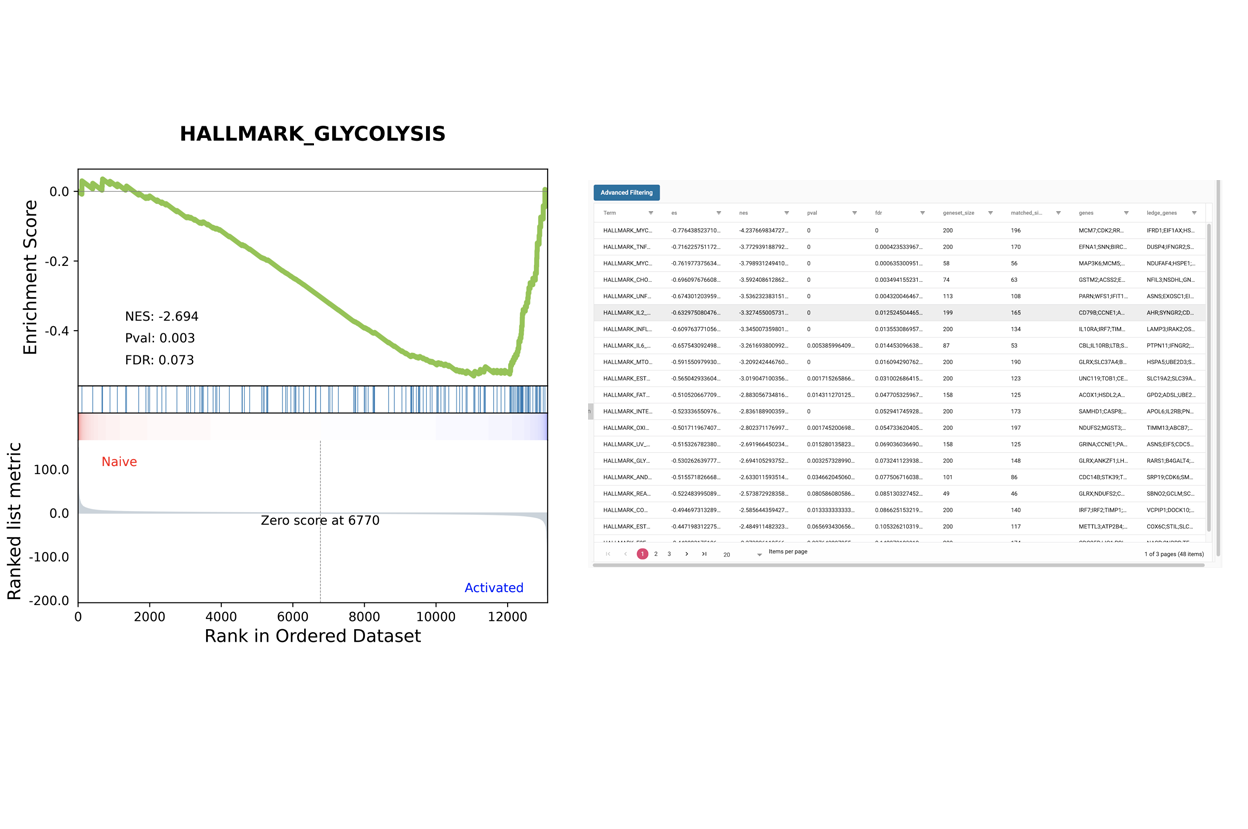
Data Analysis
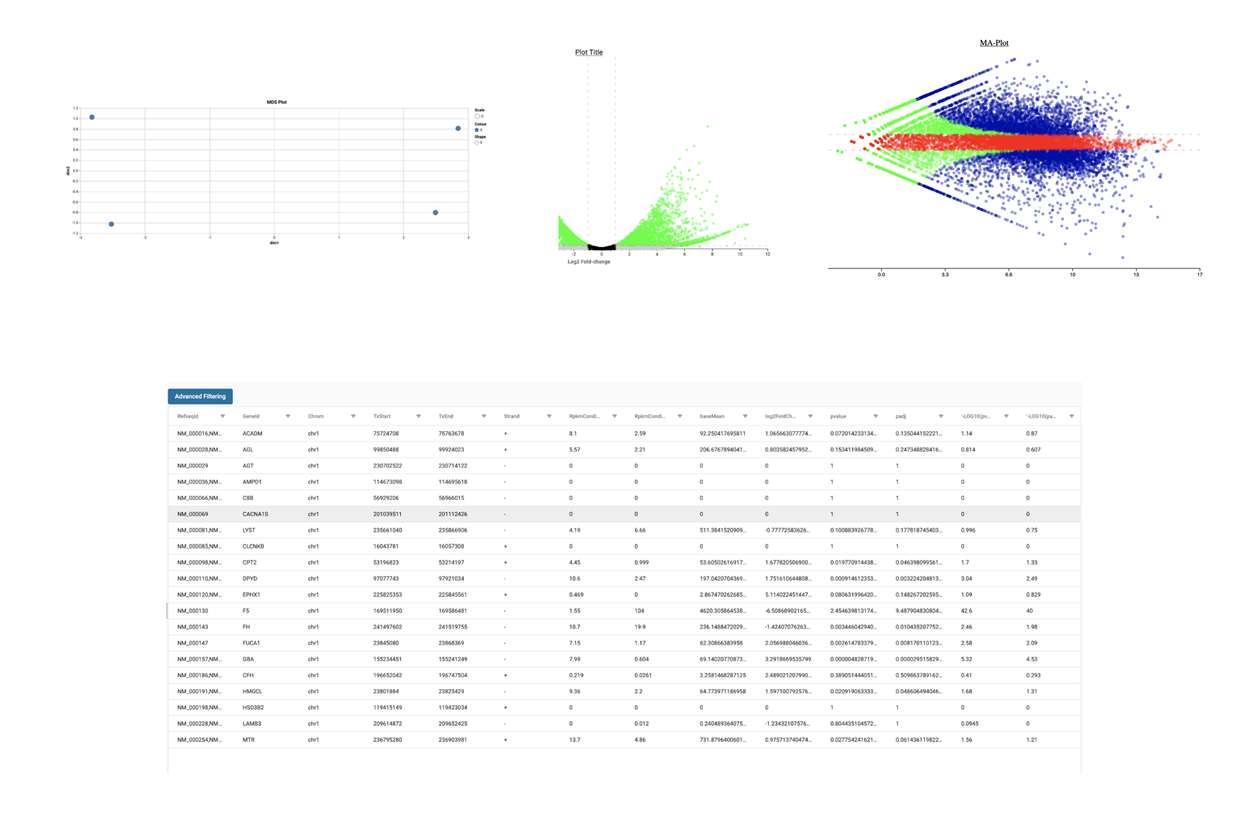
SciDAP is a no-code bioinformatics platform that enables scientists to analyze NGS-based data without a bioinformatician. It has built-in RNA-Seq pipelines based on open-source workflows, such as DESeq for differential gene expression and pipelines optimized for the dUTP method.
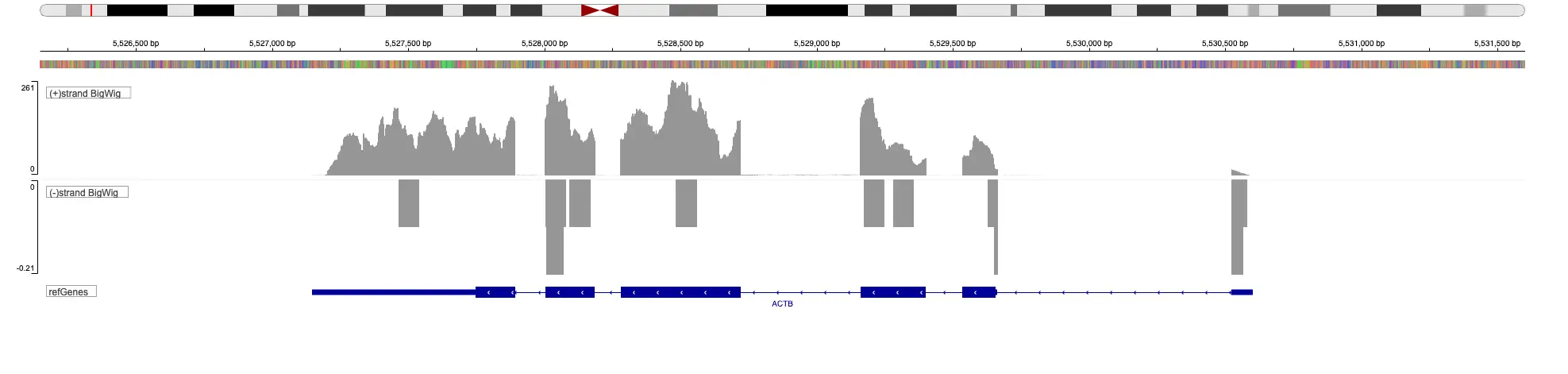
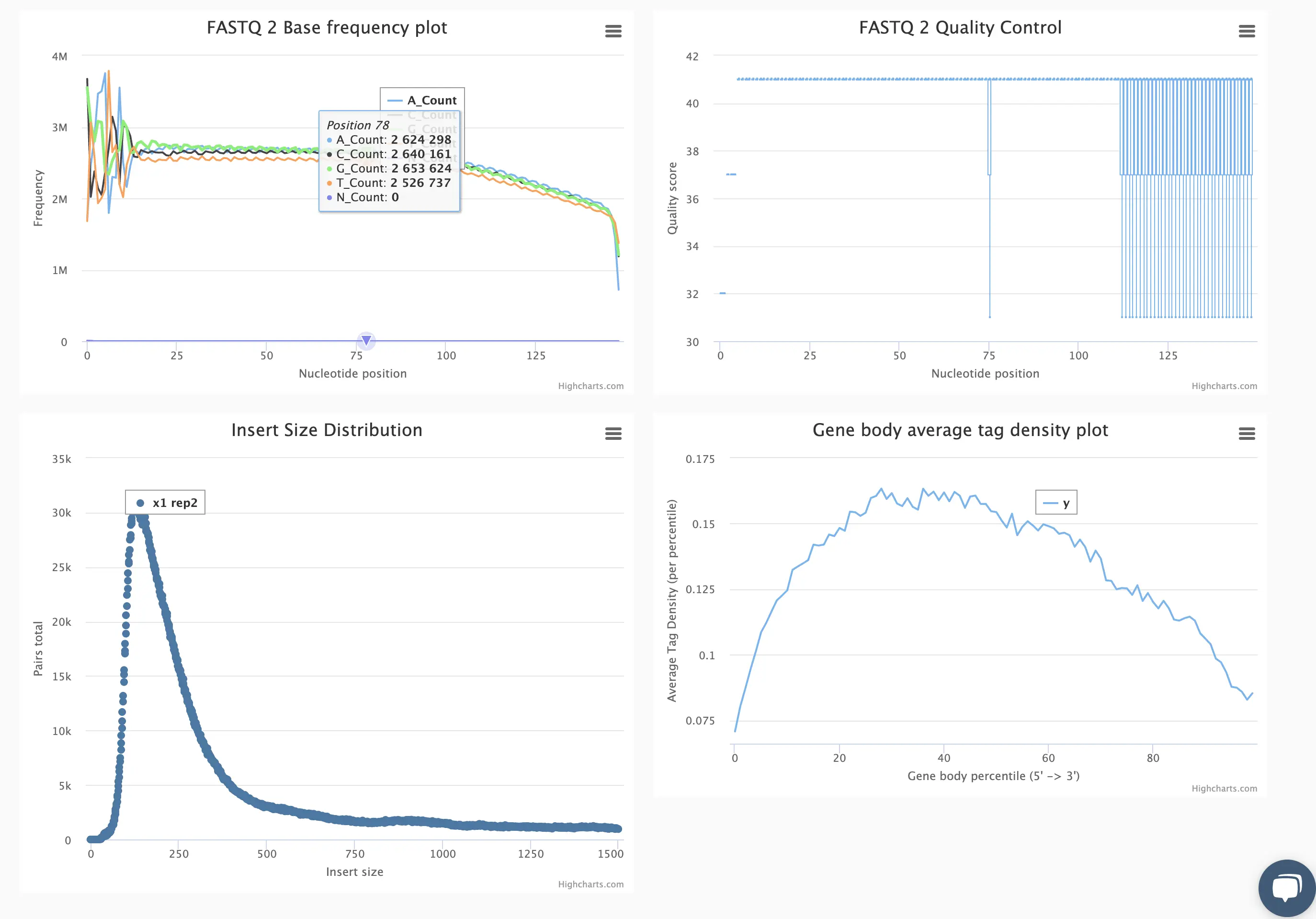
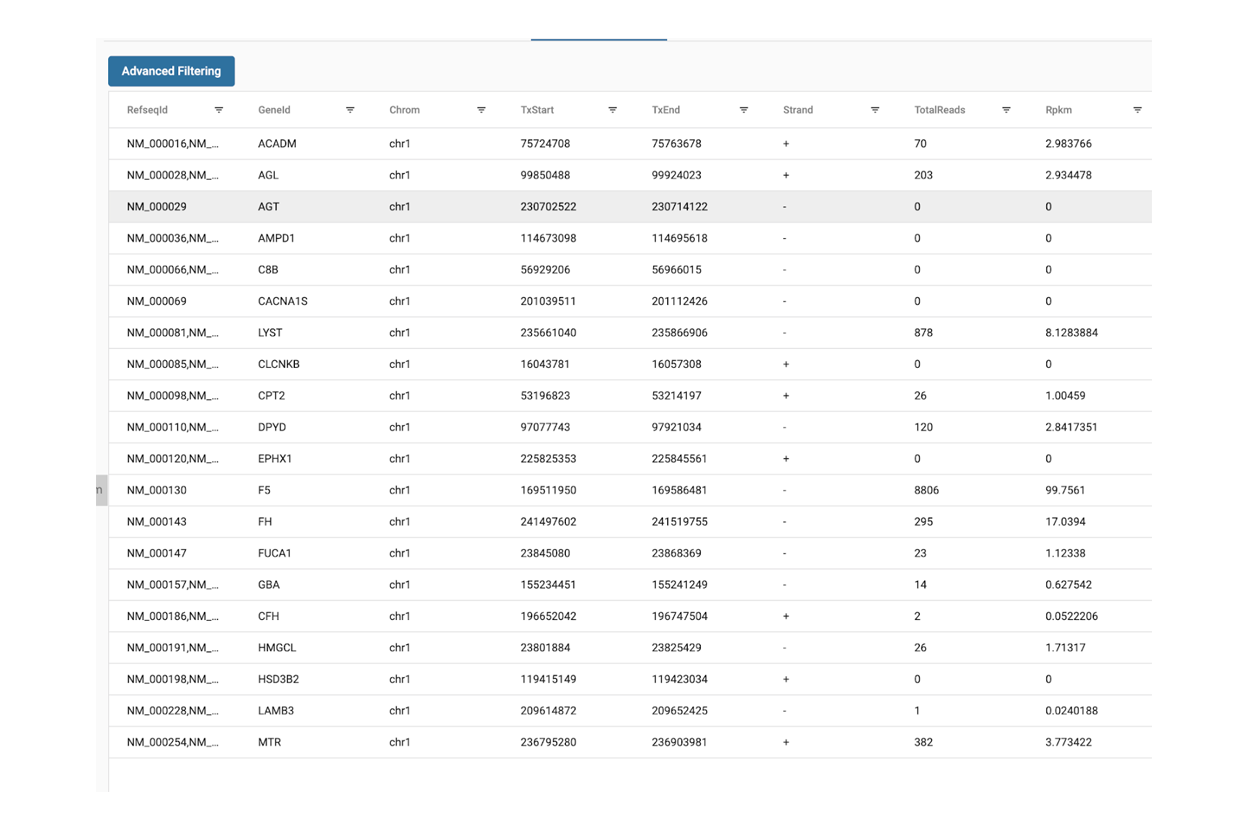
Starting with FASTQ files, analysis of RNA-Seq data begins with raw data quality control (QC) and read trimming followed by alignment of reads against a reference genome or transcriptome with RNA-STAR. Specific algorithms are applied for downstream analysis such as expression estimation, transcript isoform discovery, differential expression with DESeq or DESeq2 , GSEA analysis and other applications. Analysis ends with summarization and visualization of results.