ChIP-Seq
Overview
ChIP-Seq is a next-generation sequencing (NGS) application that analyzes DNA-protein interactions. This method is used to identify the DNA binding targets of transcription factors (TF) across the genome, revealing DNA elements regulated by a protein of interest. Histone modifications are widely assayed using ChIP-Seq and give insight into the structure and activation status of nearby genes. Many variations are available depending on the research question and material availability.
General Workflow
ChIP-Seq begins with the harvesting and fixation of the cell type of interest. Fixation using formaldehyde will cross-link the DNA with histones and trans-factors, capturing the complexes and conformations at a specific point in time. Samples are then sonicated to shear DNA fragments to be immunoprecipitated. Samples may be immunoprecipitated overnight either manually or using an automated robotic system. Both procedures will enrich the molecule of interest using antibody pulldown with protein A/G magnetic beads. Samples are de-crosslinked to release the DNA from the complexes with proteins, and DNA is amplified using a library preparation kit for NGS sequencing.
Datirium recommends the Chipmentation procedure due to its simplicity and low input requirements. Libraries can be sequenced as single or paired reads at a core facility or commercial provider. The number of reads needed for successful peak calling depends on the size of the genome, efficiency of pull-down, and the area of the genome covered by the modification or TF. Datirium recommends at least 10M for transcription factors or H3K4me3 histone mark and 50-100M for H3K27me3.
Data Analysis
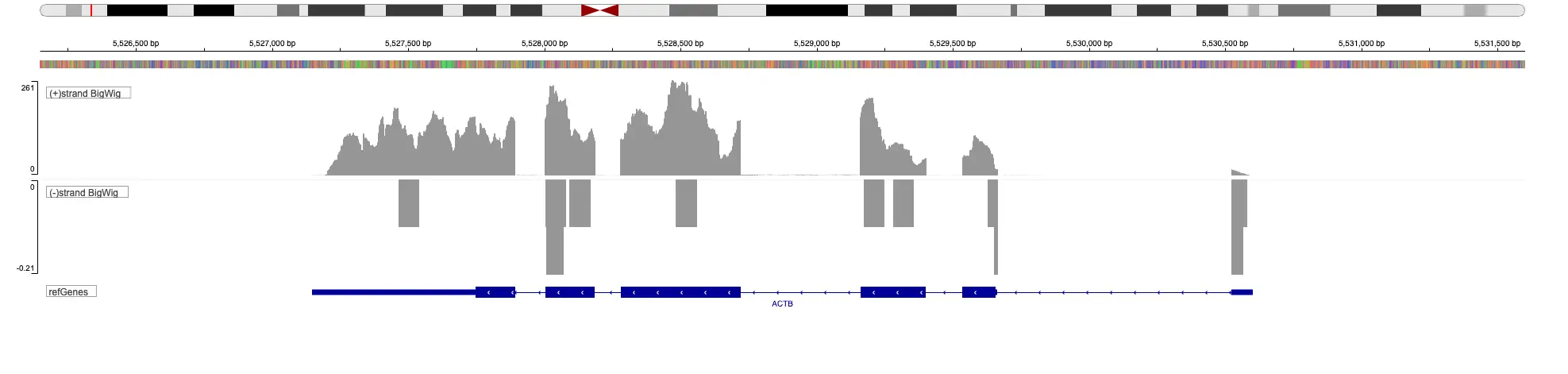
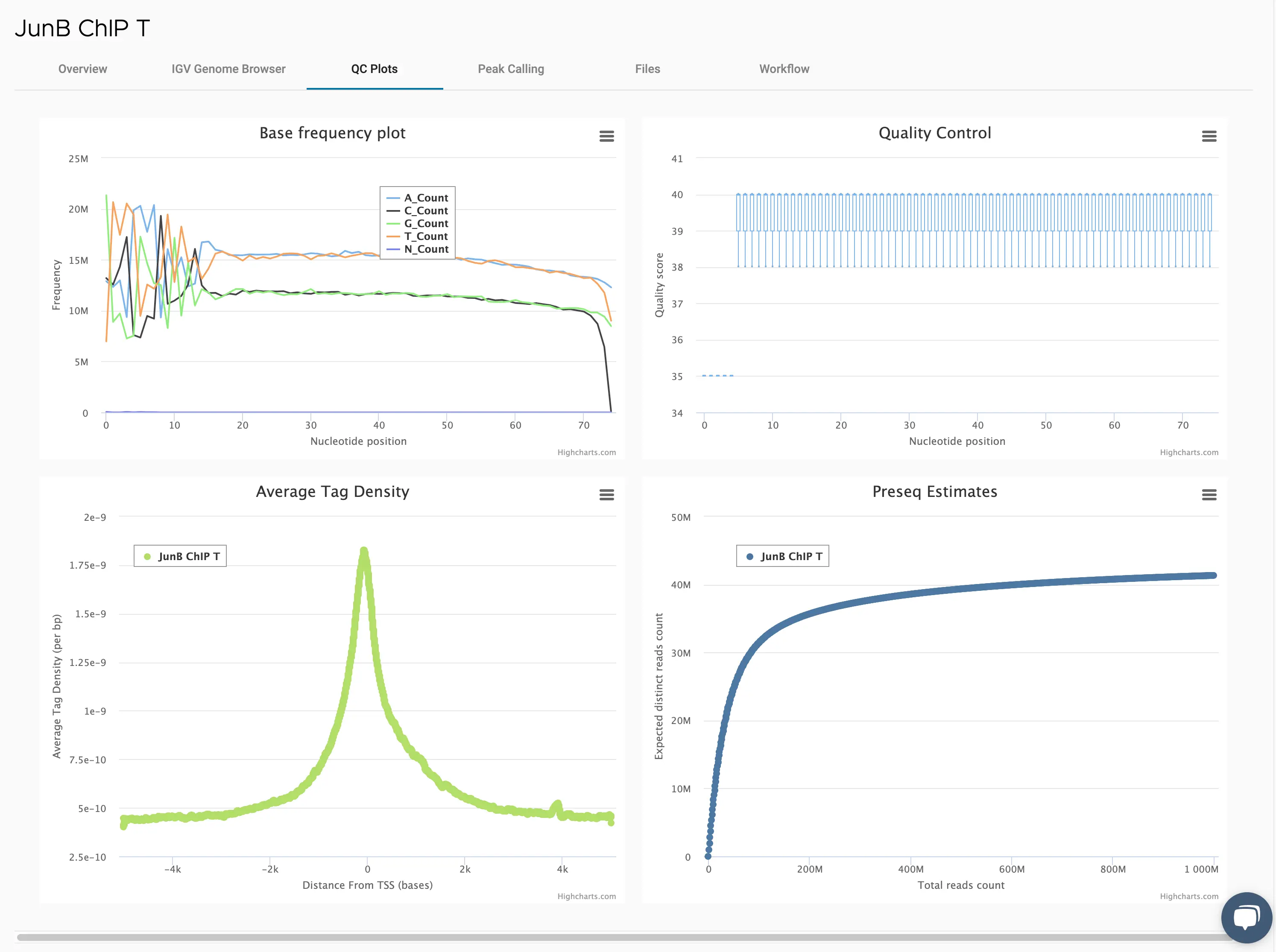
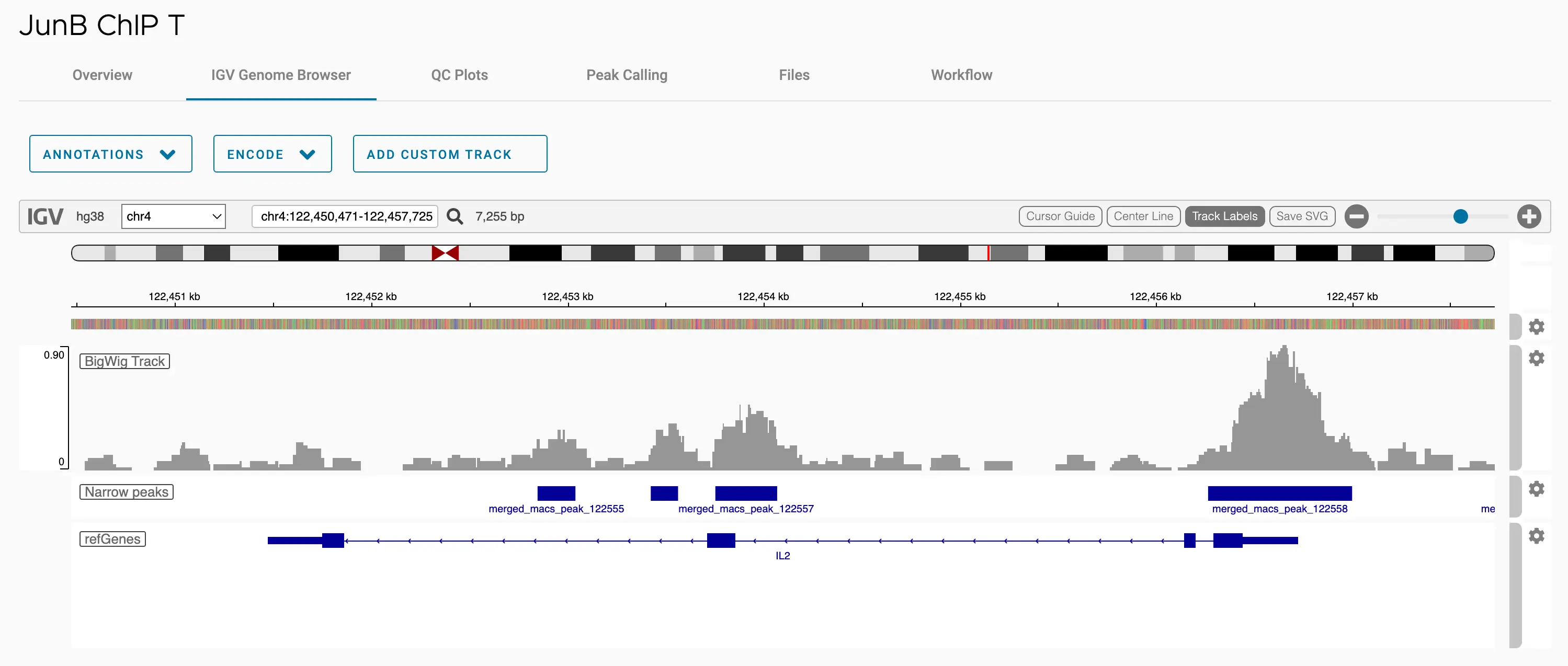
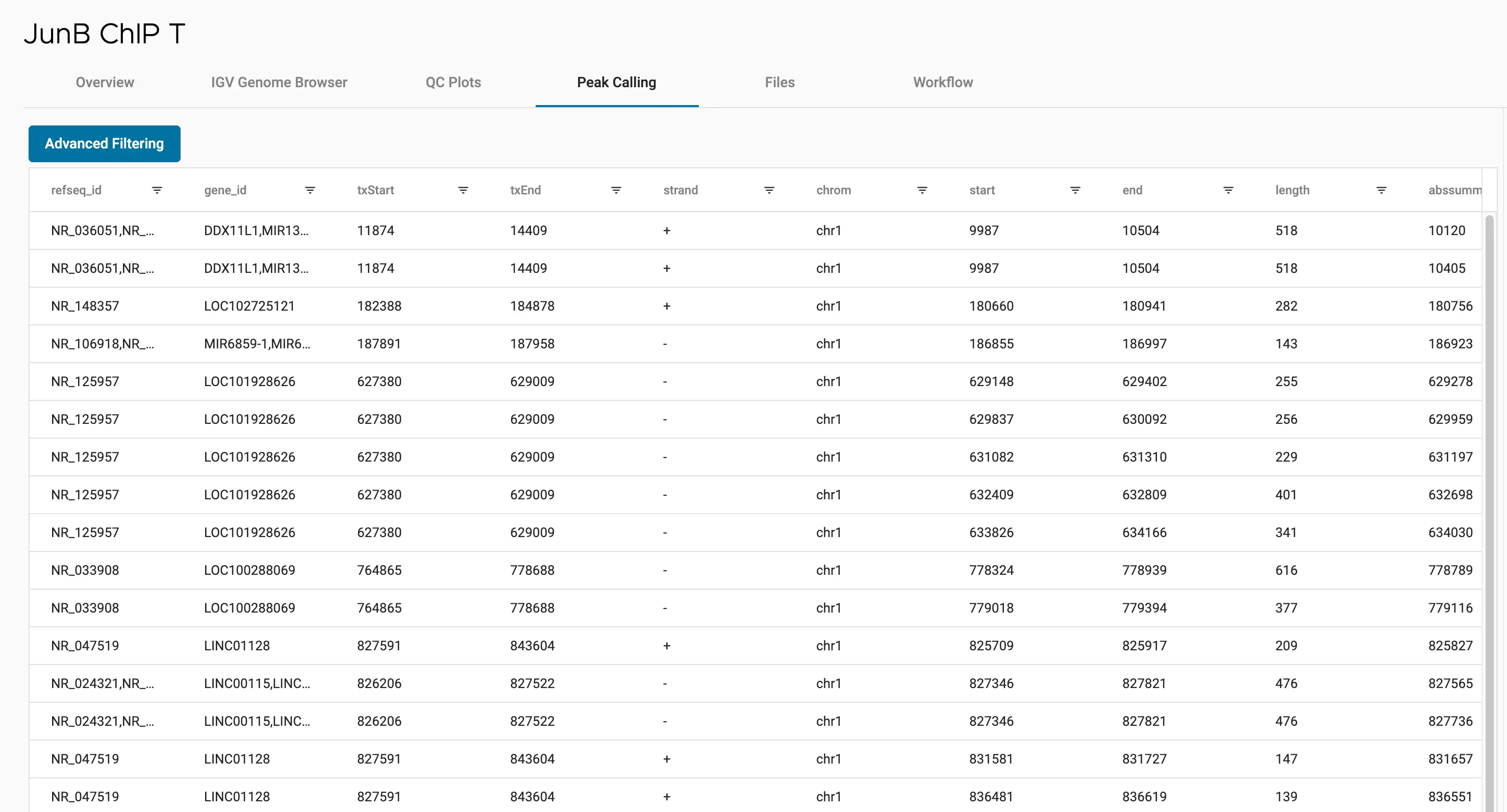
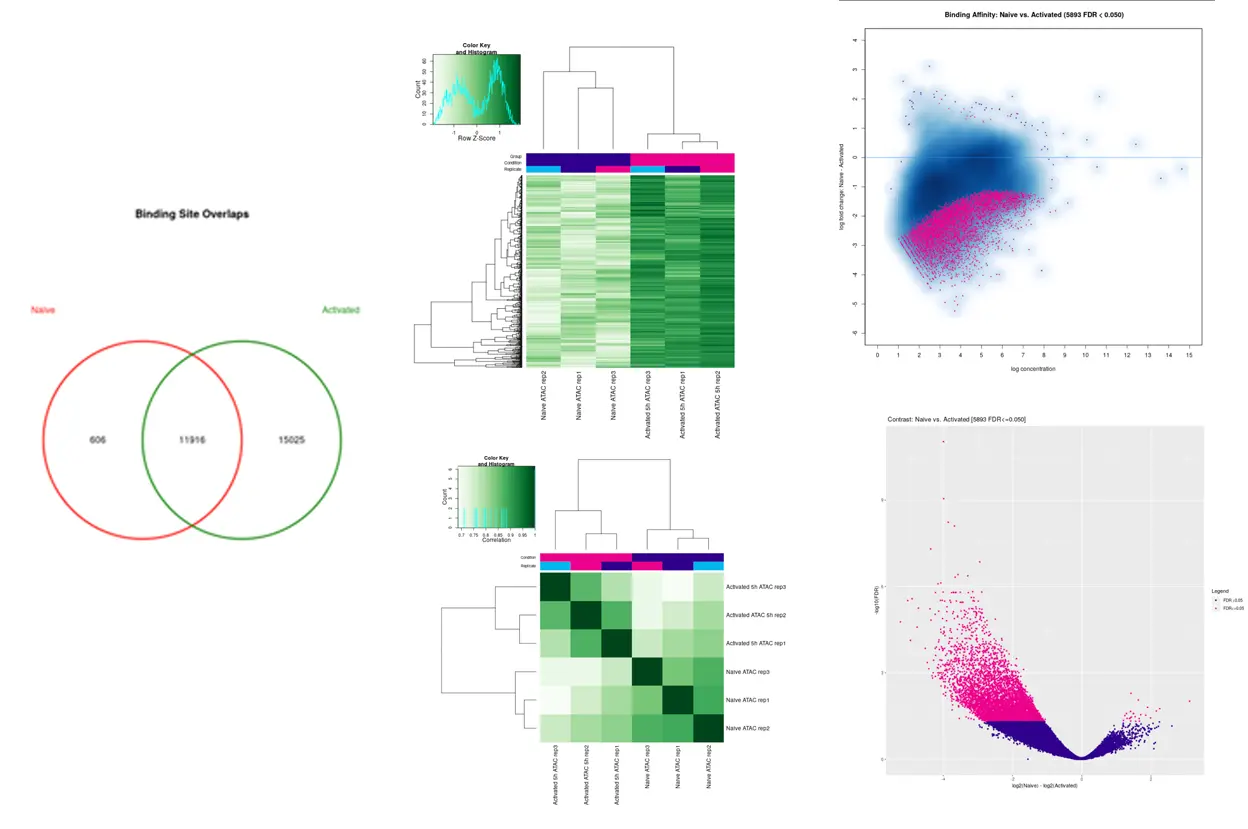
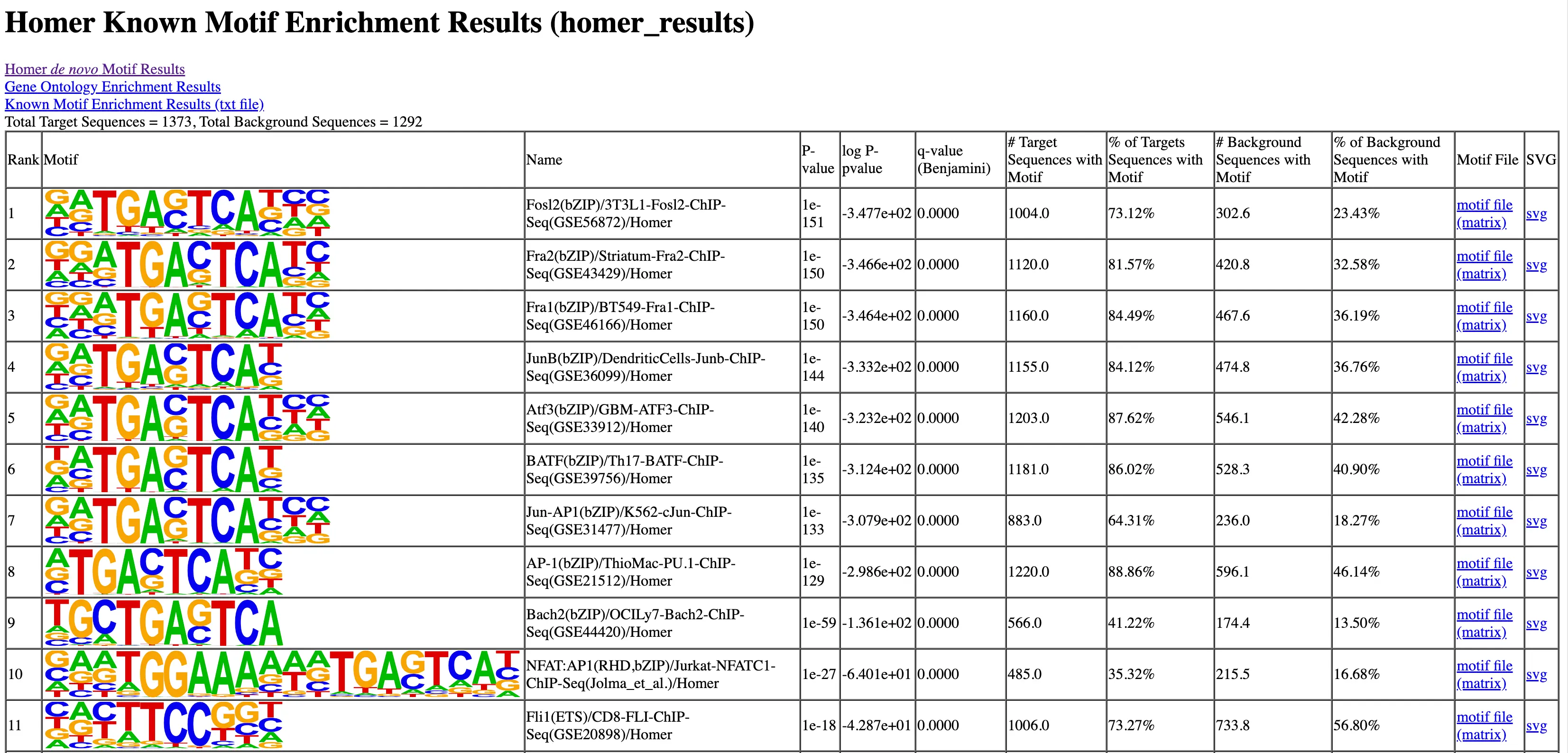
SciDAP is a no-code bioinformatics platform that enables scientists to analyze NGS-based data without a bioinformatician. It has built-in pipelines based on open-source workflows to analyze bulk data from ChIP-Seq libraries. SciDAP starts from the FASTQ files provided by most DNA core facilities and commercial service providers. Starting from raw data allows SciDAP to ensure that all experiments have been processed in the same way and simplifies the deposition of data to GEO upon publication. The data can be uploaded from the end-users’ computer, downloaded directly from an FTP server of the core facility by providing a URL, or from GEO by providing SRA accession number.