Quant-Seq
Overview
QuantSeq is a next-generation sequencing (NGS) application that offers a low-cost approach to analyzing gene expression. Instead of sequencing the entire transcript, the 3’ Tag-RNA-seq method (QuantSeq) sequences only about 50-250 bases from the 3’ end. It provides one read per transcript, instead of full coverage, which is sufficient for differential gene expression analysis, and lowers sequencing expenses.
General Workflow
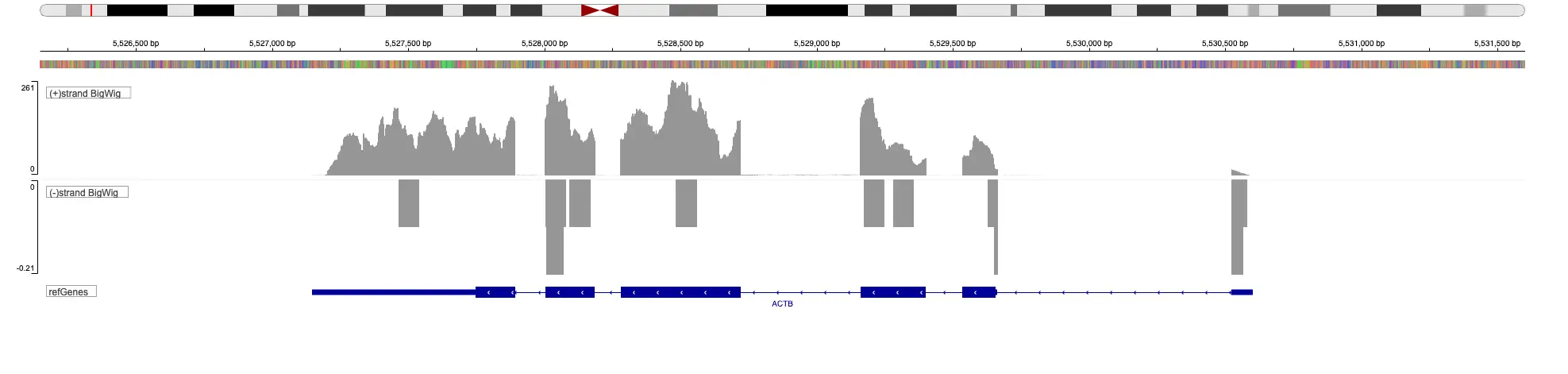
The 3’ Tag-RNA-seq method is offered as QuantSeq™ library prep kits from Lexogen. The kits provide an oligo dT primer with the Illumina-specific Read 2 linker sequence. This primer binds to all mRNA transcripts with a poly(A) tail. After first-strand cDNA synthesis and RNA removal, second-strand synthesis is initiated with a random primer and DNA polymerase to create a double-stranded DNA library. Following magnetic bead-based purification, the library is amplified and sequences required for cluster generation are introduced. NGS reads are generated towards the poly(A) tail and directly correspond to the mRNA sequence.
QuantSeq works can better accommodate degraded mRNA and FFPE samples as it only sequences the 3’ end. It is simpler and faster to make libraries compared to RNA-Seq and uses Unique Dual Indices (UDIs) to help distinguish duplicates. However, since QuantSeq does not provide full sequence of mRNA, it cannot capture splicing structures and isoforms as with RNA-Seq. And, because QuantSeq only provides reads from the 3’ end, you cannot directly compare results with RNA-Seq, but you can compare QuantSeq with mRNA-Seq libraries.
Data Analysis
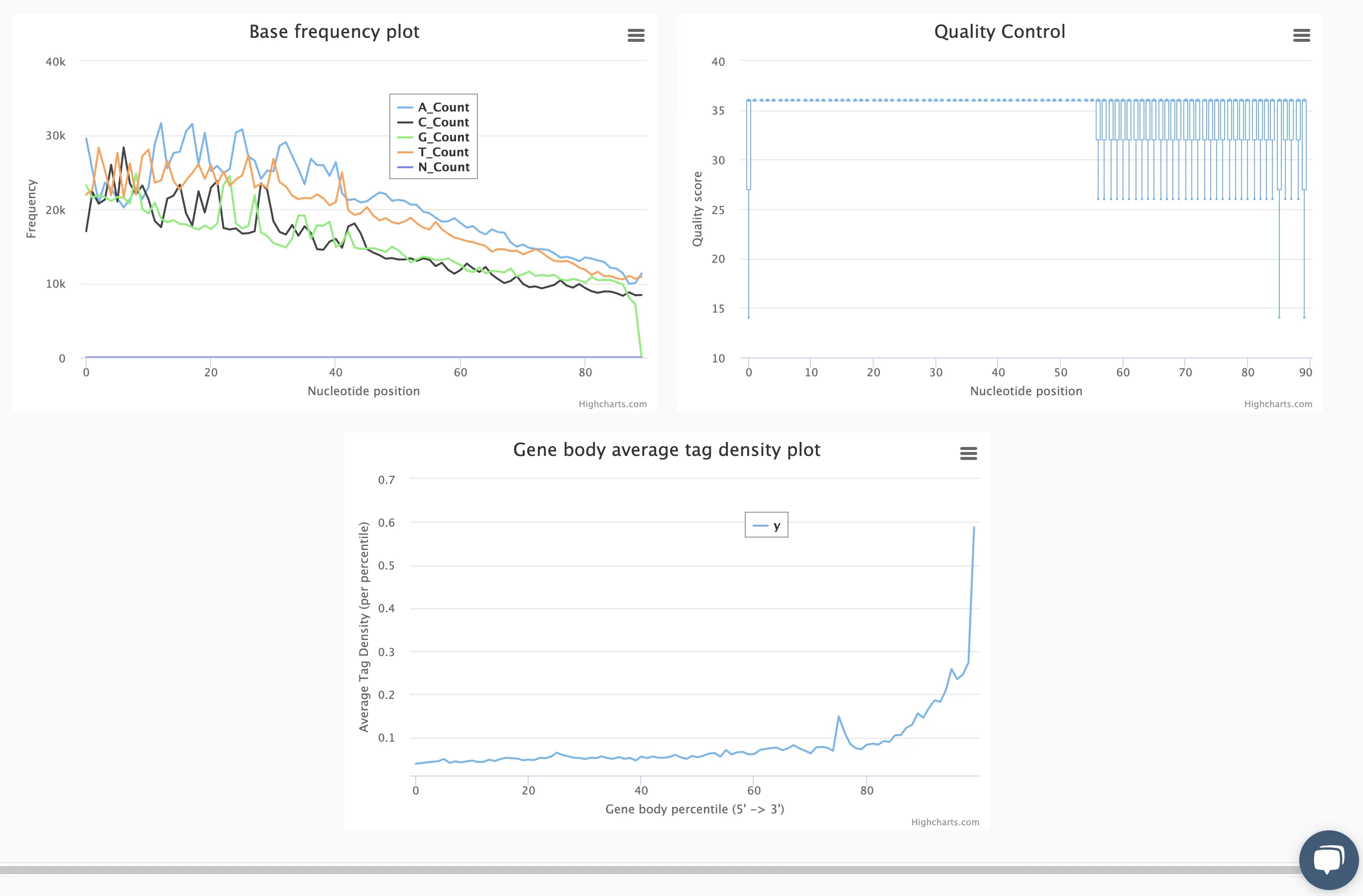
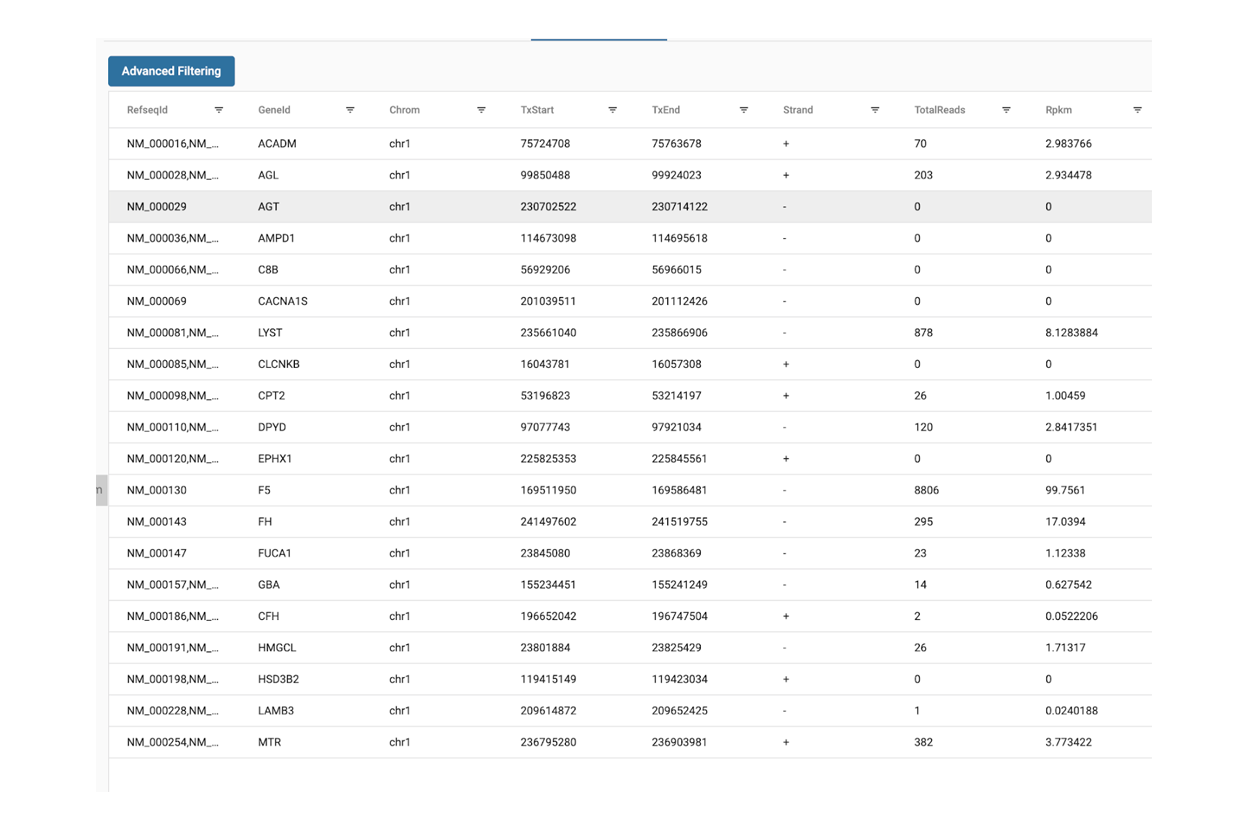
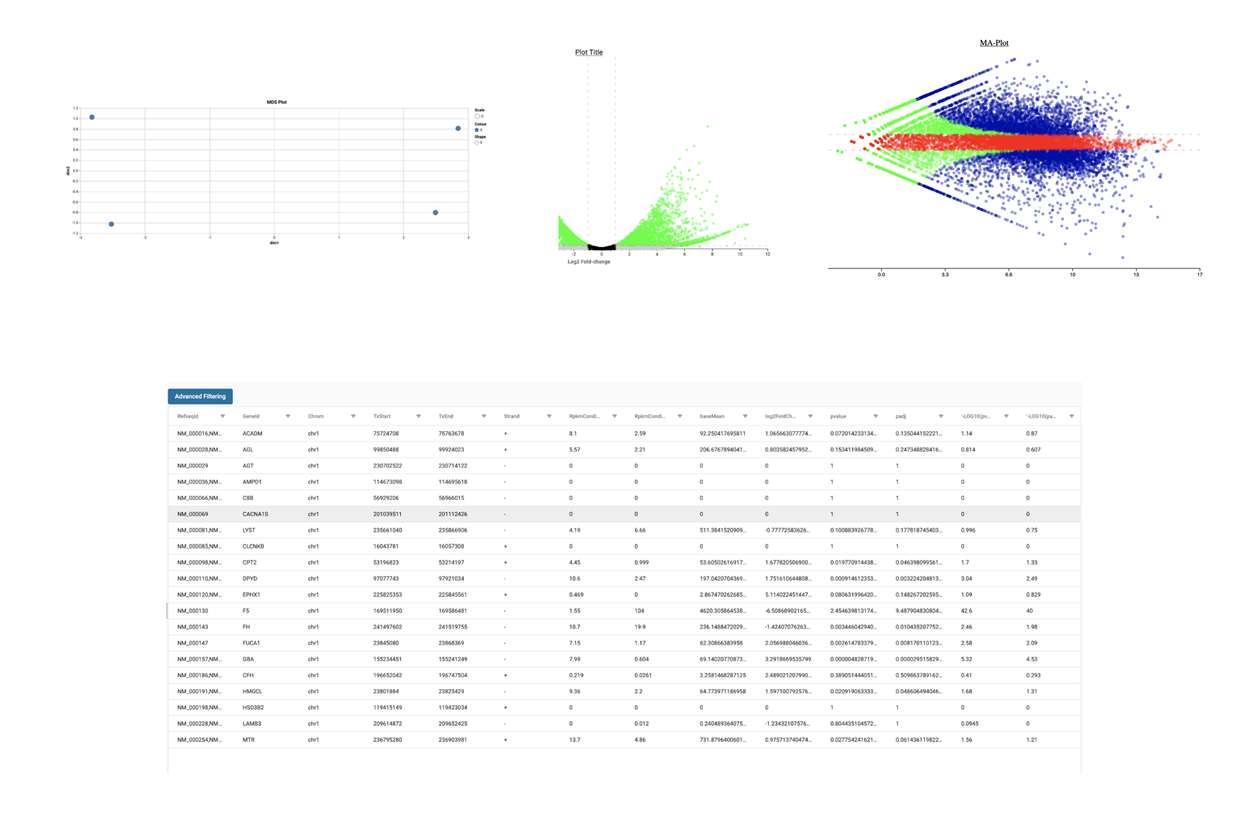
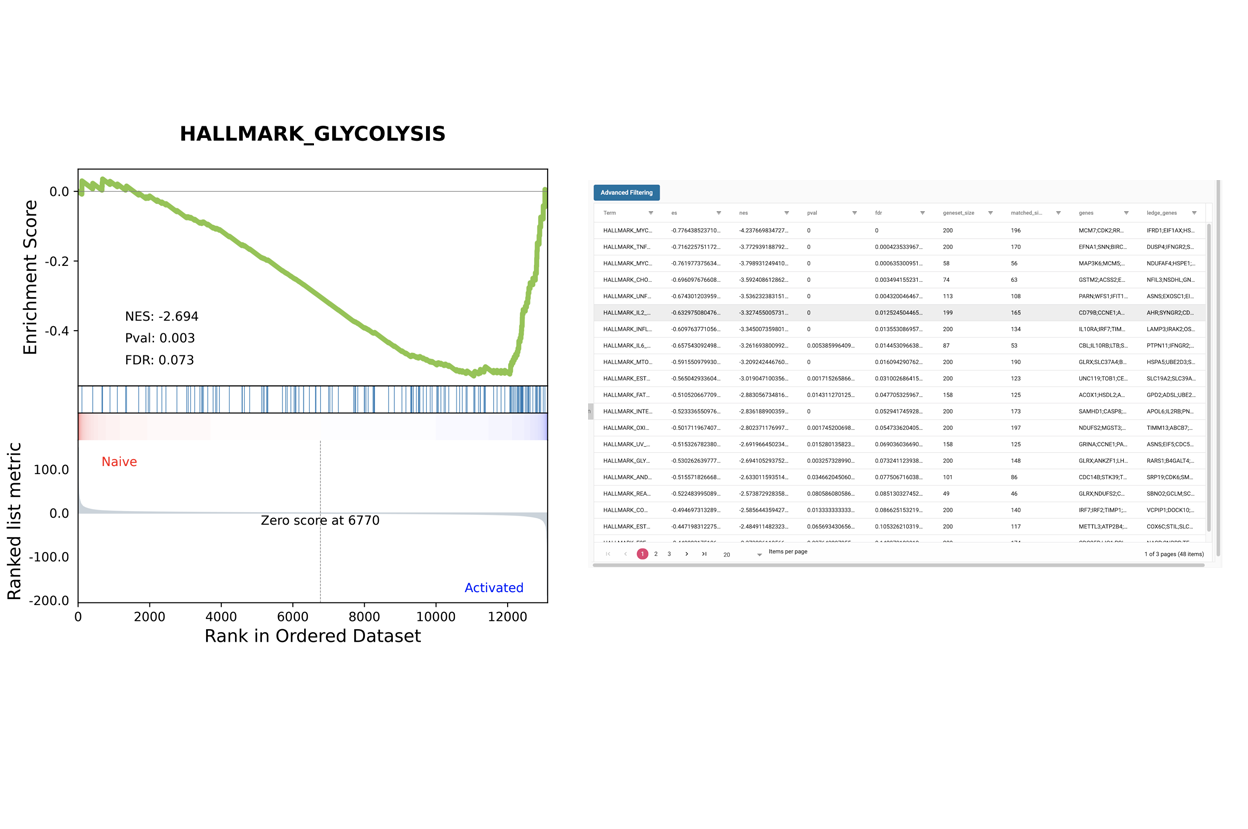
SciDAP is a no-code bioinformatics platform that enables scientists to analyze NGS-based data without a bioinformatician. It has built-in pipelines based on open-source workflows to analyze bulk data from QuantSeq™ libraries. Starting with FASTQ files, you can immediately look at differential gene expression and generate publication quality images in just a few hours. SciDAP also includes the DESeq pipeline to compare expression between two conditions, including QuantSeq and mRNA-Seq libraries. The outcome is a table that contains the read number for each gene.